I am a postdoctoral researcher at the Eric and Wendy Schmidt Center of the Broad Institute of MIT and Harvard. My research is at the intersection of computer science, molecular biology, and statistics. I develop algorithms and machine learning models to integrate multi-modal genomic data, with a goal of understanding cellular dynamics in health and disease and assisting cell state engineering. I received my Ph.D. in computer science and computational biology from Brown University, under the advisement of Ritambhara Singh, Ph.D. (primary advisor) and Sorin Istrail, Ph.D. My doctoral dissertation was on optimal transport algorithms for integrated analysis of single-cell multi-omics data.
Since January 2024, I co-chair the Models, Inference, and Algorithms (MIA) seminar series at the Broad, where we host hybrid (in-person & online) talks by computational scientists in bio(medicine). You can check out our upcoming schedule and past talks.
Apart from research, I have had the opportunity to work on various fun projects that can be found here. Outside of research, I enjoy boardgames with friends, swimming, hiking, playing with my cat, and practicing the ukulele and violin.
Feel free to e-mail me if you’d like to talk.
Research Interests
Methodology
Application areas
- Representation learning
- Optimal transport
- Manifold learning
- Bayesian statistics and inference
- Variable selection
- Deep learning
- Causality
- Graph algorithms
- Regulatory genomics
- Functional genomics
- Single-cell sequencing & imaging
- Multi-omics
- 3D genome
- Precision medicine
- Structural biology
Education
| 2018 - 2023 |
Ph.D. in Computer Science and Computational Biology
(3.90/4.00)
M.Sc. in Computer Science
(4.00/4.00)
Brown University (Providence, RI)
|
| 2013 - 2017 |
B.Sc. in Engineering (with concentration in Bioengineering)
(3.67/4.00)
Olin College of Engineering (Needham, MA)
|
| 2008 - 2013 |
TEVITOL High School with IB Diploma (Gebze, Turkey)
|
Professional Experience
| July 2023 - Present |
Broad Institute of MIT and Harvard, EWSC Postdoctoral Fellow (Cambridge, MA) |
| June 2022 - Aug 2022 |
Microsoft Research, Research Intern (Redmond, WA) |
| June 2020 - Sep 2020 |
Microsoft Research, Research Intern (Redmond, WA) |
| June 2017 - August 2018 |
Massachusetts Institute of Technology, Research Support Associate (Cambridge, MA) |
| Jan 2016 - Oct 2016 |
Design That Matters, Student Engineer (Salem, MA) |
| Jan 2015 - Dec 2015 |
Daktari Diagnostics, Student Engineer (Cambridge, MA) |
2024
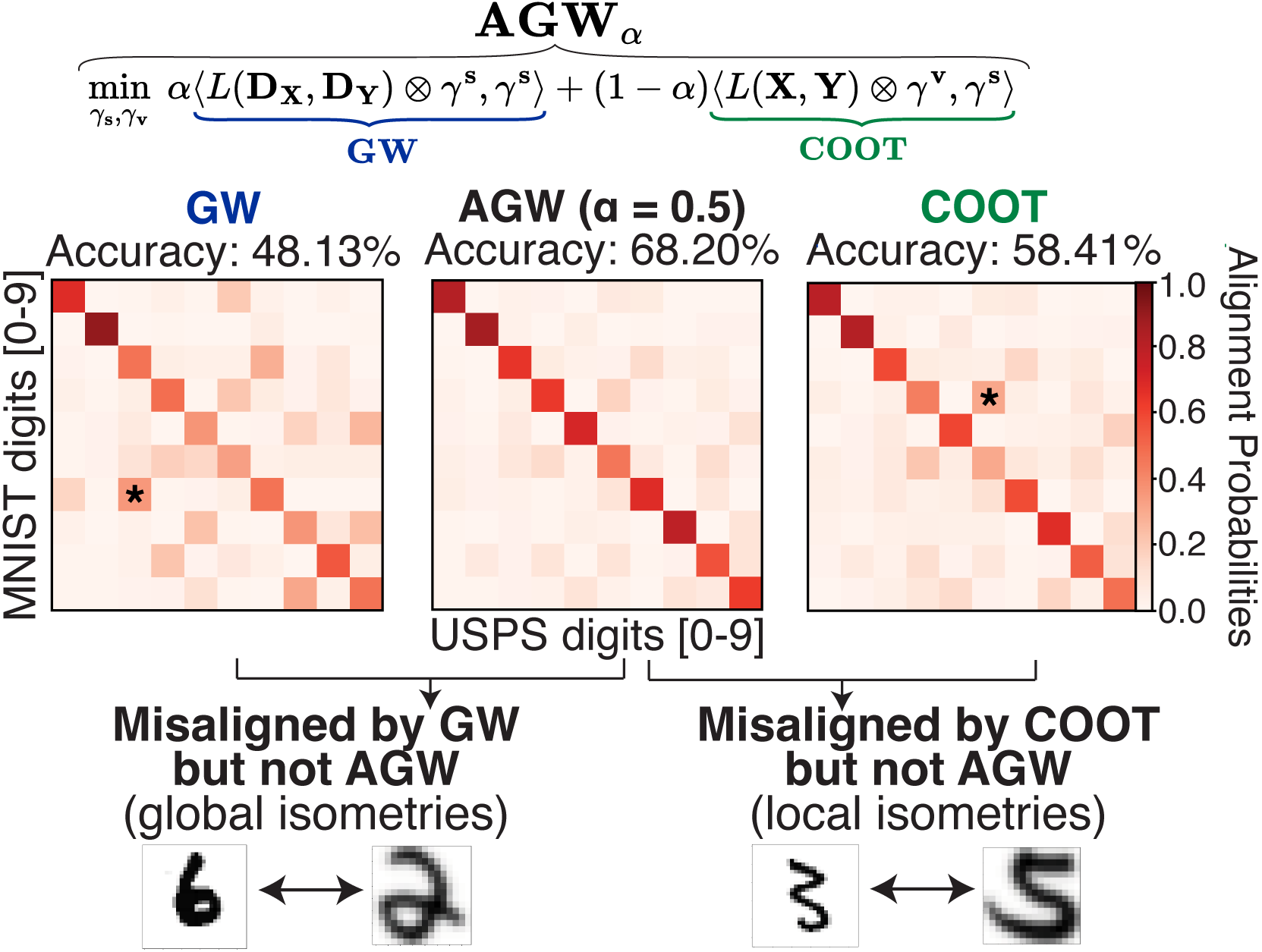 |
Breaking isometric ties and introducing priors in Gromov-Wasserstein distances
P. Demetci, Q.H. Tran, I. Redko, R. Singh
Proceedings of the 27th International Conference on Artificial Intelligence and Statistics
(AISTATS 2024)
Proceedings of Machine Learning Research (PMLR) -- to appear
[11]
[abstract] [paper] [code]
Gromov-Wasserstein distance has many applications in machine learning due to its ability to compare measures across metric spaces and its invariance to isometric transformations. However, in certain applications, this invariant property can be too flexible, thus undesirable. Moreover, the Gromov-Wasserstein distance solely considers pairwise sample similarities in input datasets, disregarding the raw feature representations. We propose a new optimal transport formulation, called Augmented Gromov-Wasserstein (AGW), that allows for some control over the level of rigidity to transformations. It also incorporates feature alignments, enabling us to better leverage prior knowledge on the input data for improved performance. We present theoretical insights into the proposed method. We then demonstrate its usefulness for single-cell multi-omic alignment tasks and heterogeneous domain adaptation in machine learning.
|
2023
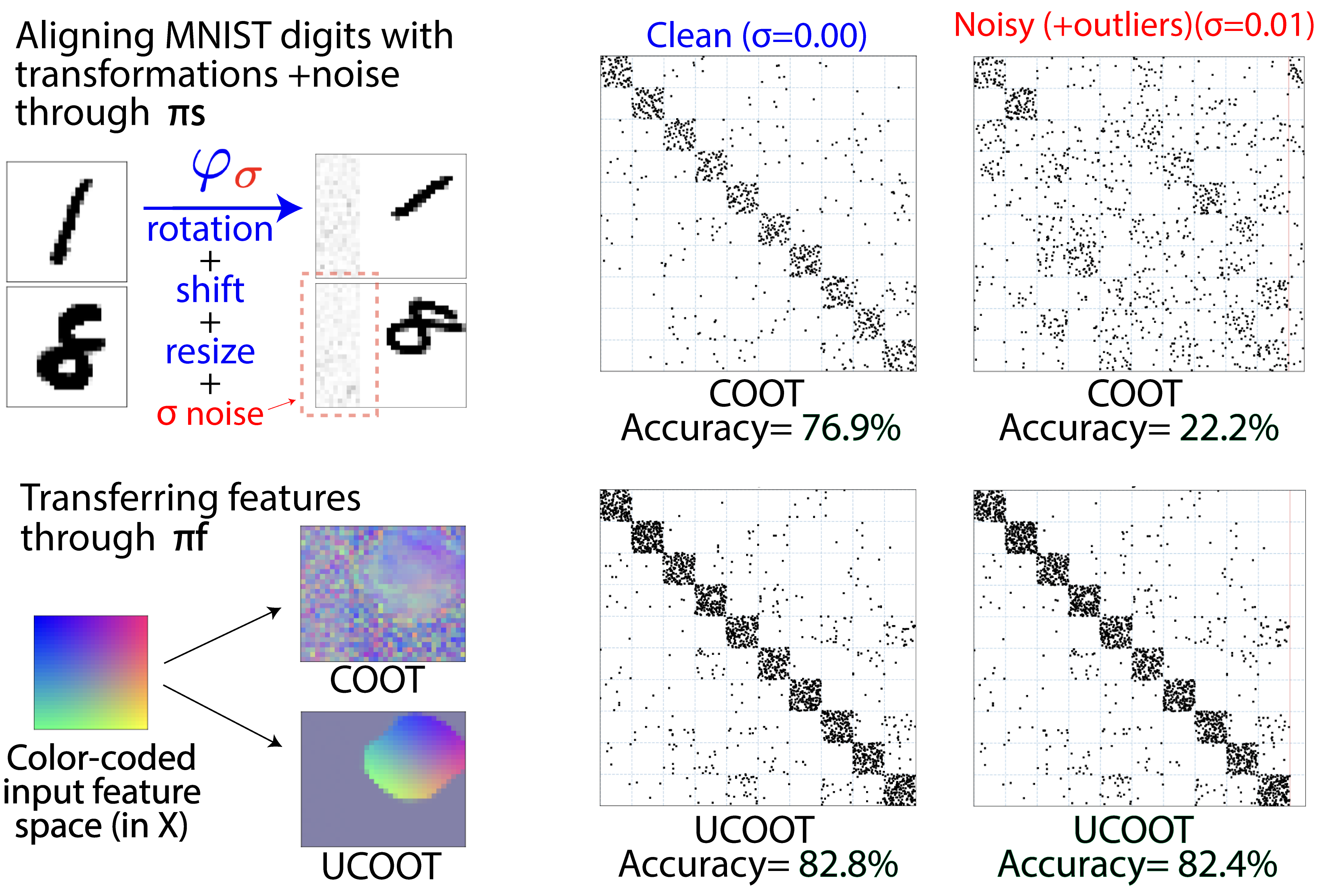 |
Unbalanced CO-Optimal Transport
Q.H. Tran, H. Janati, N. Courty, R. Flamary, I. Redko, P. Demetci, R Singh
Proceedings of the 37th AAAI Conference on Artificial Intelligence
(AAAI 2023)
[10]
[abstract] [paper] [code]
Optimal transport (OT) compares probability distributions by computing a meaningful alignment between their samples. CO-optimal transport (COOT) takes this comparison further by inferring an alignment between features as well. While this approach leads to better alignments and generalizes both OT and Gromov-Wasserstein distances, we provide a theoretical result showing that it is sensitive to outliers that are omnipresent in real-world data. This prompts us to propose unbalanced COOT for which we provably show its robustness to noise in the compared datasets. To the best of our knowledge, this is the first such result for OT methods in incomparable spaces. With this result in hand, we provide empirical evidence of this robustness for the challenging tasks of heterogeneous domain adaptation with and without varying proportions of classes and simultaneous alignment of samples and features across single-cell measurements.
|
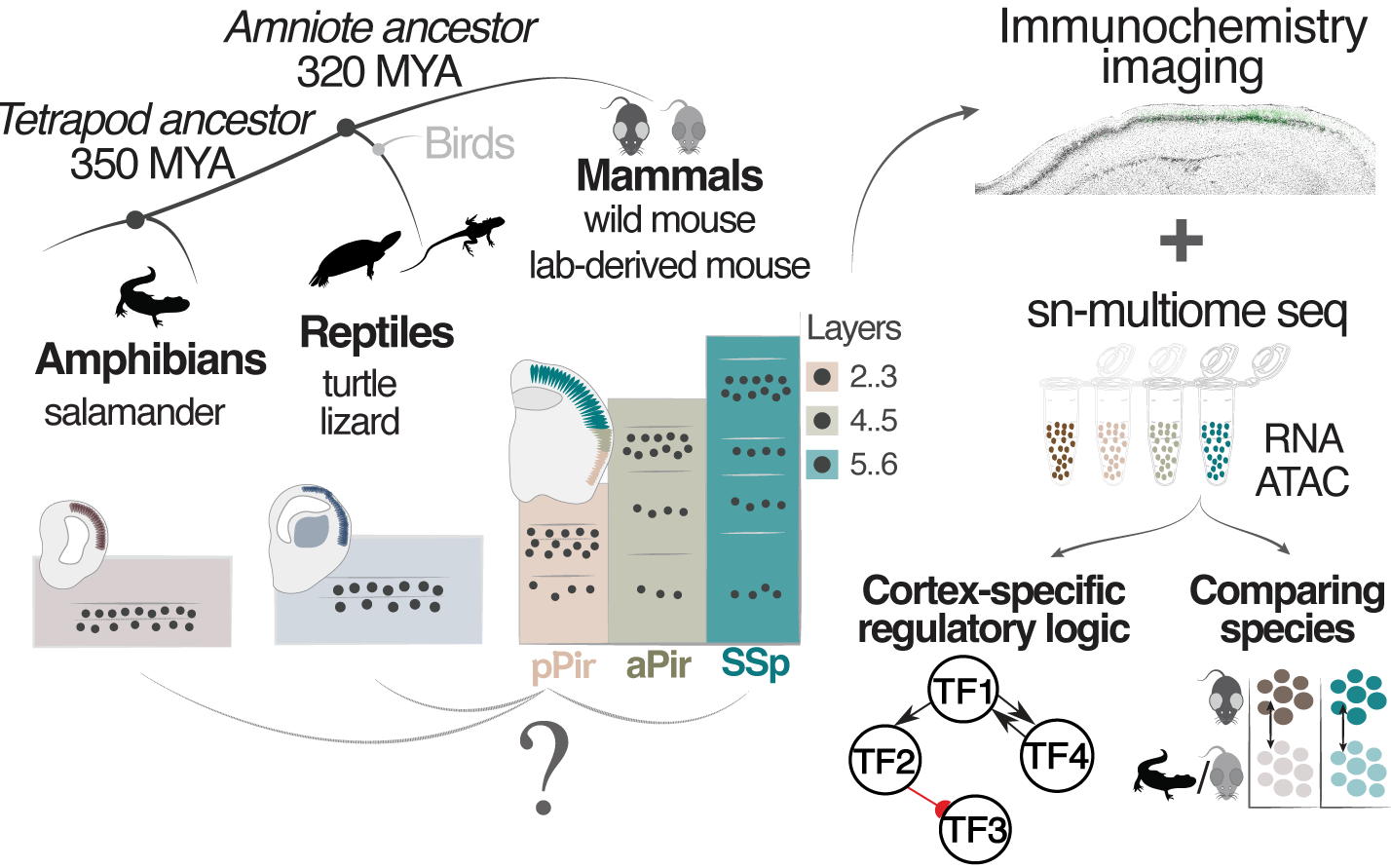 |
Mammalian olfactory cortex neurons retain molecular signatures of ancestral cell types
S. Zeppilli, A. Ortega Gurrola, P. Demetci, DH. Brann, R. Attey, N. Zilkha, T. Kimchi, SR. Datta, R. Singh, MA. Tosches, A. Crombach, A. Fleischmann
bioRxiv (Under review at Nature Neuroscience)
[9]
[abstract] [paper] [code]
The cerebral cortex diversified extensively during vertebrate evolution. Intriguingly, the three-layered mammalian olfactory cortex resembles the cortical cytoarchitecture of non-mammals yet evolved alongside the six-layered neocortex, enabling unique comparisons for investigating cortical neuron diversification. We performed single-nucleus multiome sequencing across mouse three- to six-layered cortices and compared neuron types across mice, reptiles and salamander. We identified neurons that are olfactory cortex-specific or conserved across mouse cortical areas. However, transcriptomically similar neurons exhibited area-specific epigenetic states. Additionally, the olfactory cortex showed transcriptomic divergence between lab and wild-derived mice, suggesting enhanced circuit plasticity through adult immature neurons. Finally, olfactory cortex neurons displayed marked transcriptomic similarities to reptile and salamander neurons. Together, these data indicate that the mammalian olfactory cortex retains molecular signatures representative of ancestral cortical traits.
|
2022
 |
Unsupervised Integration of Single-Cell Multi-omics Datasets with Disproportionate Cell-Type Representation
P. Demetci, R. Santorella, B. Sandstede, R. Singh
Proceedings of the 26th Annual Intl. Conference on Research in Computational Molecular Biology
(RECOMB 2022)
Springer Nature Lecture Notes in Bioinformatics (2022) pp 3-19
[8]
[abstract] [paper] [code]
Integrated analysis of multi-omics data allows the study of how different molecular views in the genome interact to regulate cellular processes; however, with a few exceptions, applying multiple sequencing assays on the same single cell is not possible. While recent unsupervised algorithms align single-cell multi-omic datasets, these methods have been primarily benchmarked on co-assay experiments rather than the more common single-cell experiments taken from separately sampled cell populations. Therefore, most existing methods perform subpar alignments on such datasets. Here, we improve our previous work Single Cell alignment using Optimal Transport (SCOT) by using unbalanced optimal transport to handle disproportionate cell-type representation and differing sample sizes across single-cell measurements. We show that our proposed method, SCOTv2, consistently yields quality alignments on five real-world single-cell datasets with varying cell-type proportions and is computationally tractable. Additionally, we extend SCOTv2 to integrate multiple (M ≥ 2) single-cell measurements and present a self-tuning heuristic process to select hyperparameters in the absence of any orthogonal correspondence information.
|
2021
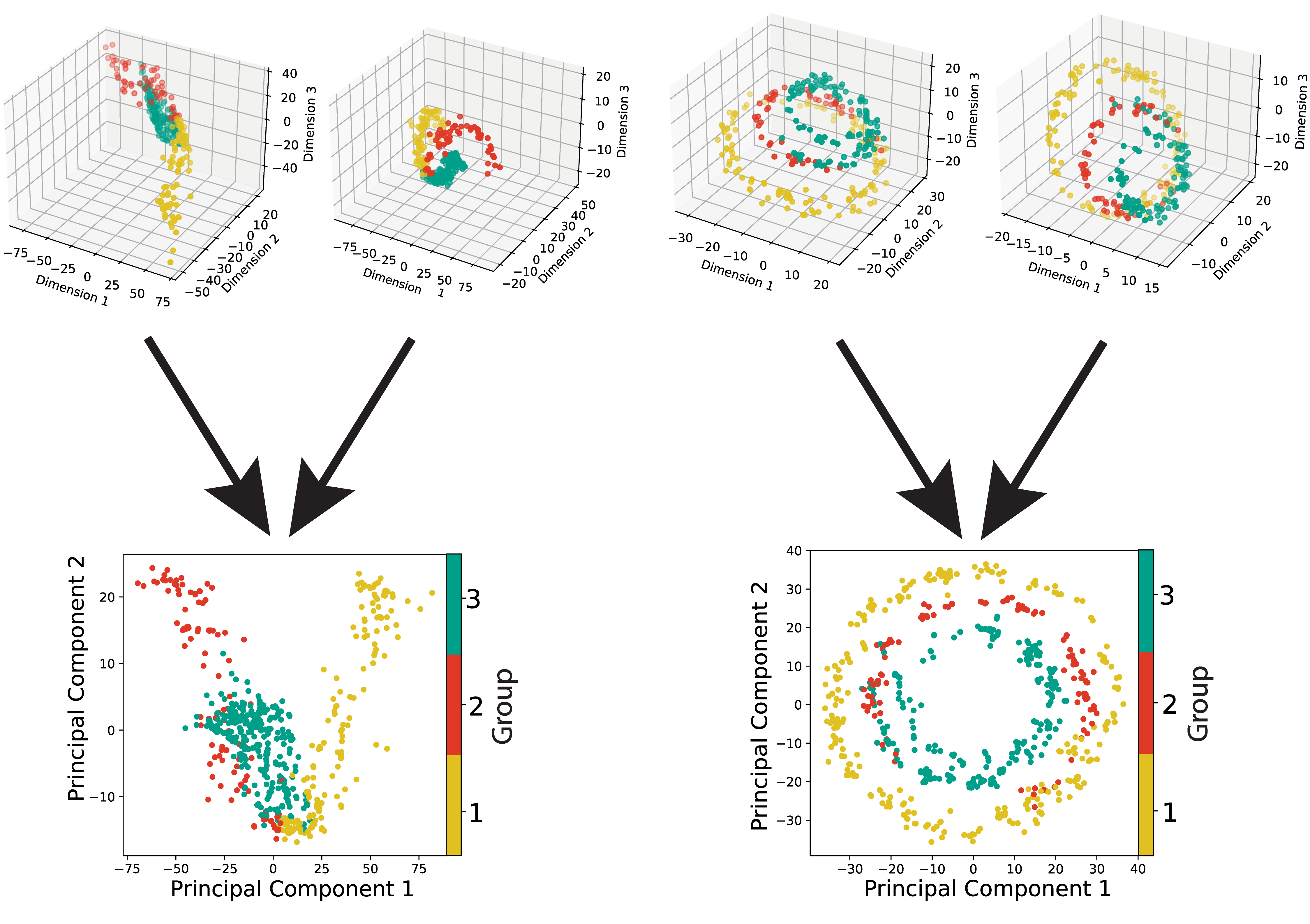 |
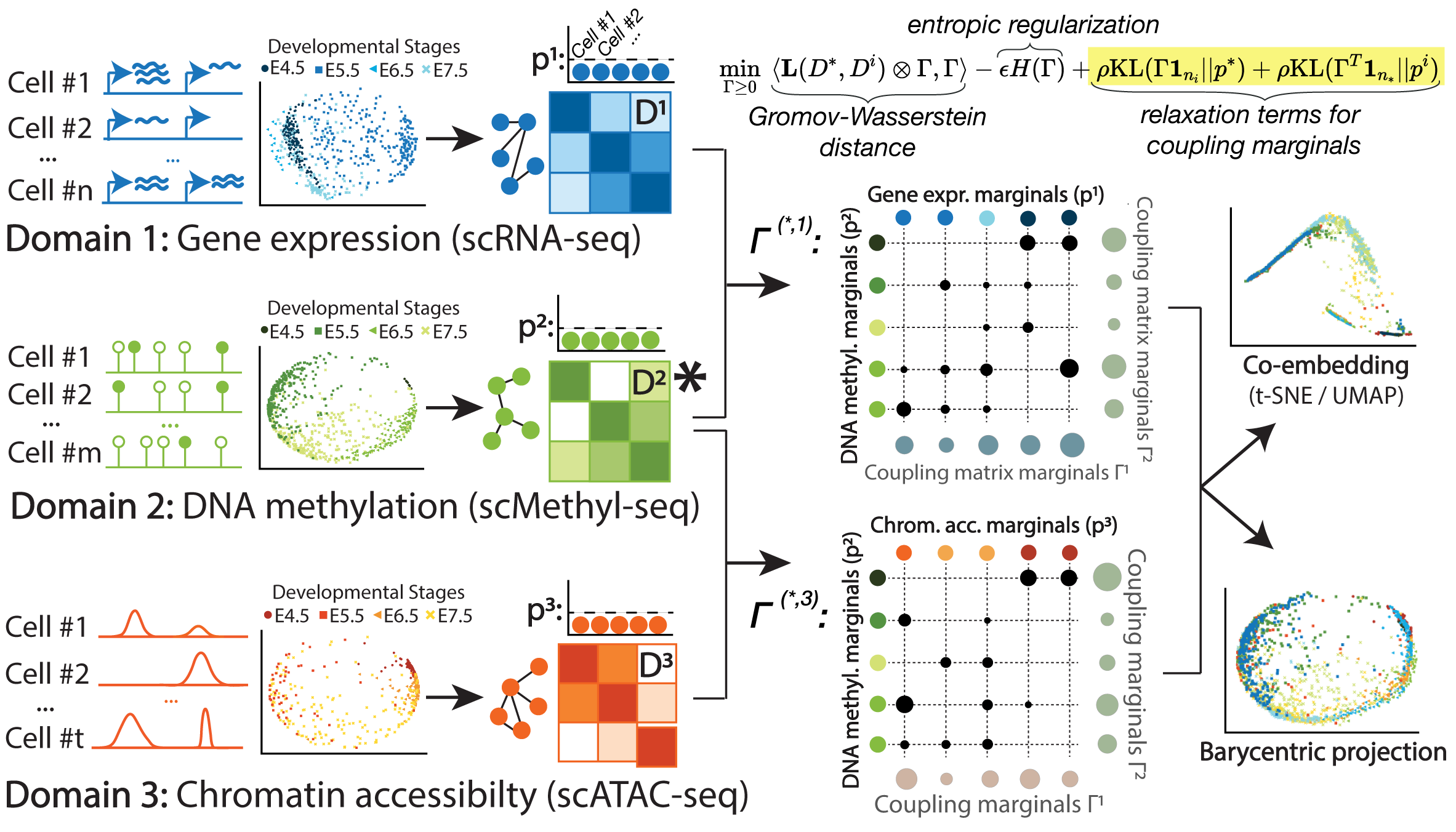
SCOT: Single-cell multi-omics integration with optimal transport
P. Demetci*, R. Santorella*, B. Sandstede, W. Stafford Noble and Ritambhara Singh#
*Equal Contribution, #Corresponding Author
Journal of Computational Biology, 2021
[7]
[abstract] [paper] [code] [tutorial]
Recent advances in sequencing technologies have allowed us to capture various aspects of the genome at single-cell resolution. However, with the exception of a few of co-assaying technologies, it is not possible to simultaneously apply different sequencing assays on the same single cell. In this scenario, computational integration of multi-omic measurements is crucial to enable joint analyses. This integration task is particularly challenging due to the lack of sample-wise or feature-wise correspondences. We present Single-Cell alignment with Optimal Transport (SCOT), an unsupervised algorithm that uses Gromov-Wasserstein optimal transport to align single-cell multi-omics datasets. SCOT performs on par with the current state-of-the-art unsupervised alignment methods, is faster, and requires tuning of fewer hyperparameters. More importantly, SCOT uses a self-tuning heuristic to guide hyperparameter selection based on Gromov-Wasserstein distance. Thus, in the fully unsupervised setting, SCOT aligns single-cell datasets better than the existing methods without requiring any orthogonal correspondence information. </a>
|
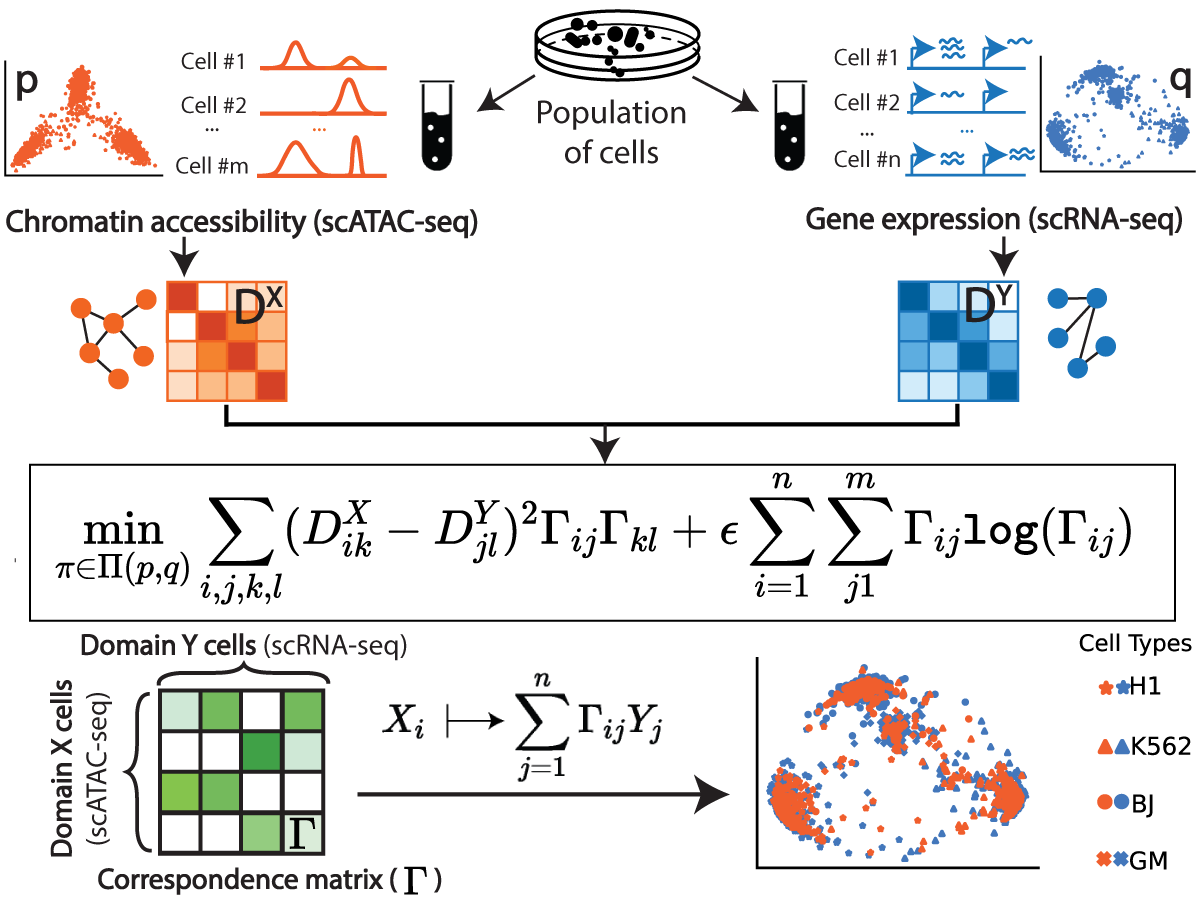 |
Gromov-Wasserstein Optimal Transport to Align Single-Cell Multi-Omics Data
P. Demetci*, R. Santorella*, B. Sandstede, W. Stafford Noble and Ritambhara Singh#
*Equal Contribution, #Corresponding Author
International Conference on Research in Computational Molecular Biology
(RECOMB 2021)
[6]
[abstract] [paper] [code] [tutorial]
ICML WCB Best Poster Award
Data integration of single-cell measurements is critical for our understanding of cell development and disease, but the lack of correspondence between different types of single-cell measurements makes such efforts challenging. Several unsupervised algorithms are capable of aligning heterogeneous types of single-cell measurements in a shared space, enabling the creation of mappings between single cells in different data modalities.
We present Single-Cell alignment using Optimal Transport (SCOT), an unsupervised learning algorithm that uses Gromov Wasserstein-based optimal transport to align single-cell multi-omics datasets. SCOT calculates a probabilistic coupling matrix that matches cells across two datasets. The optimization uses k-nearest neighbor graphs, thus preserving the local geometry of the data. We use the resulting coupling matrix to project one single-cell dataset onto another via a barycentric projection. We compare the alignment performance of SCOT with state-of-the-art algorithms on three simulated and two real datasets. Our results demonstrate that SCOT yields results that are comparable in quality to those of competing methods, but SCOT is significantly faster and requires tuning fewer hyperparameters. The code is available at https://github.com/rsinghlab/SCOT
|
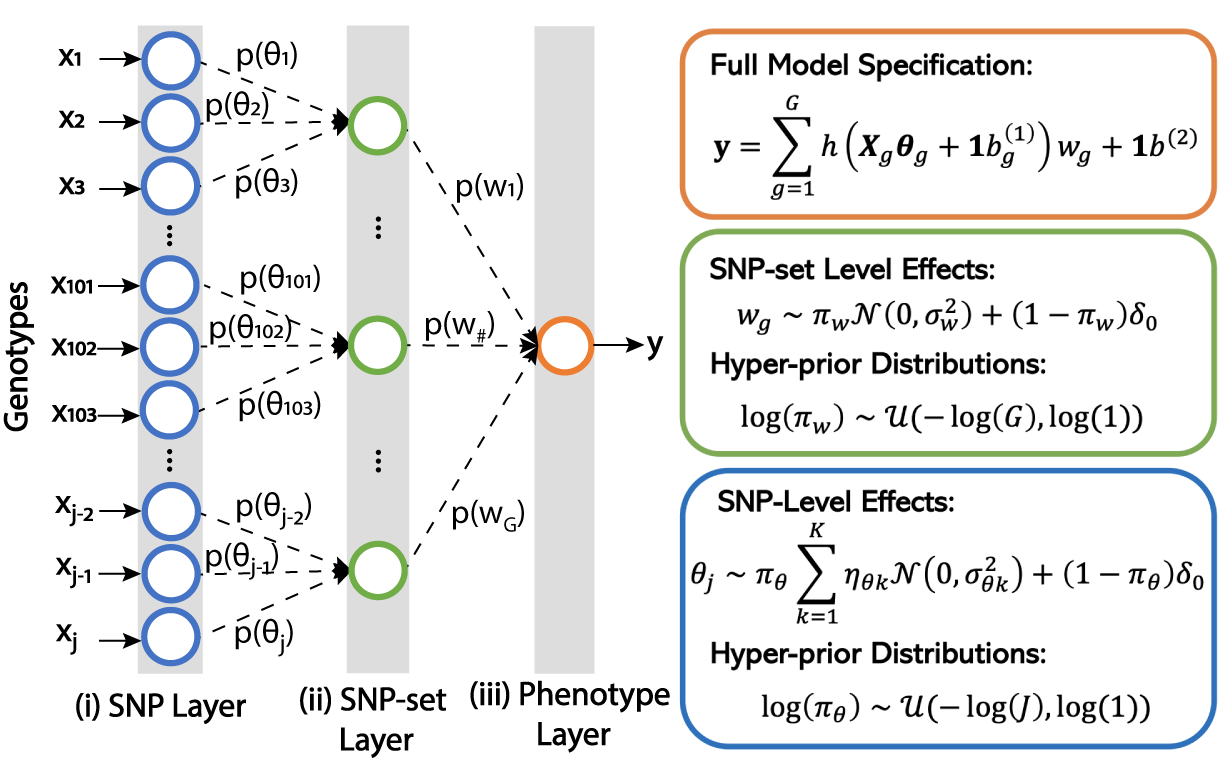 |
Multi-scale Inference of Genetic Trait Architecture using Biologically Annotated Neural Networks
P. Demetci,W. Cheng,Gregory Darnell, Xiang Zhou, Sohini Ramachandran, Lorin Crawford#
PLOS Genetics, 2021
[5]
[abstract] [paper]
In this article, we present Biologically Annotated Neural Networks (BANNs), a nonlinear probabilistic framework for association mapping in genome-wide association (GWA) studies. BANNs are feedforward models with partially connected architectures that are based on biological annotations. This setup yields a fully interpretable neural network where the input layer encodes SNP-level effects, and the hidden layer models the aggregated effects among SNP-sets. We treat the weights and connections of the network as random variables with prior distributions that reflect how genetic effects manifest at different genomic scales. The BANNs software uses variational inference to provide posterior summaries which allow researchers to simultaneously perform (i) mapping with SNPs and (ii) enrichment analyses with SNP-sets on complex traits. Through simulations, we show that our method improves upon state-of-the-art association mapping and enrichment approaches across a wide range of genetic architectures. We then further illustrate the benefits of BANNs by analyzing real GWA data assayed in approximately 2,000 heterogenous stock of mice from the Wellcome Trust Centre for Human Genetics and approximately 7,000 individuals from the Framingham Heart Study. Lastly, using a random subset of individuals of European ancestry from the UK Biobank, we show that BANNs is able to replicate known associations in high and low-density lipoprotein cholesterol content.</a>
|
2020
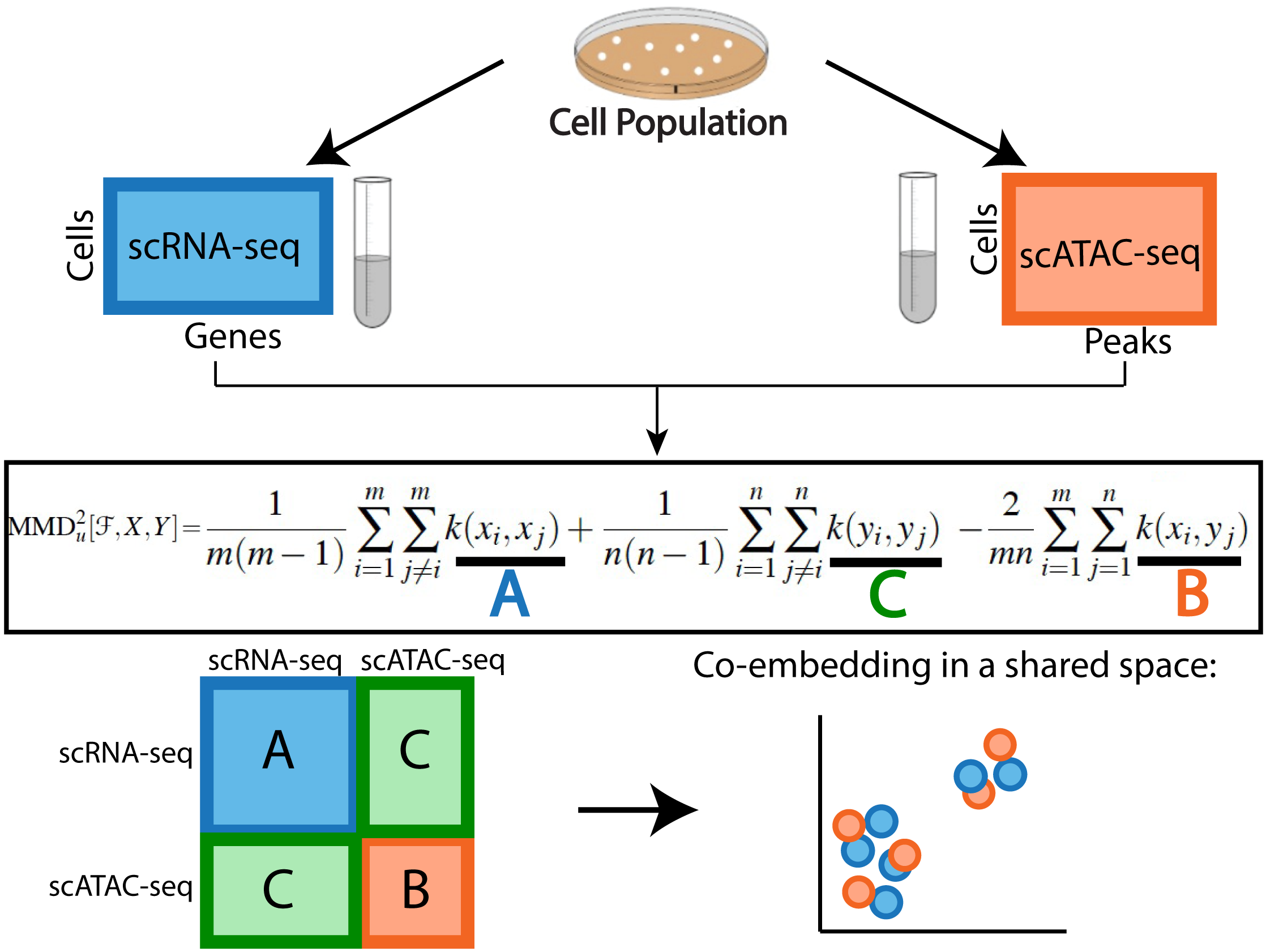 |
Unsupervised Manifold Alignment for Single-Cell Multi-Omics Data
R. Singh#, P. Demetci, G. Bonora, V. Ramani, C. Lee, H. Fang, Z. Duan, X. Deng, J. Shendure, C. Disteche and W. Stafford Noble#
#Corresponding Authors
Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics
(ACM BCB 2020)
[4]
[abstract] [paper] [code]
Integrating single-cell measurements that capture different properties of the genome is vital to extending our understanding of genome biology.This task is challenging due to the lack of a shared axis across datasets obtained from different types of single-cell experiments. For most such datasets, we lack corresponding information among the cells (samples) and the measurements (features). In this scenario, unsupervised algorithms that are capable of aligning single-cell experiments are critical to learning an in silico co-assay that can help draw correspondences among the cells.Maximum mean discrepancy-based manifold alignment (MMD-MA) is such an unsupervised algorithm. Without requiring correspondence information, it can align single-cell datasets from different modalities in a common shared latent space, showing promising results on simulations and a small-scale single-cell experiment with 61 cells.However, it is essential to explore the applicability of this method to larger single-cell experiments with thousands of cells so that it can be of practical interest to the community.In this paper, we apply MMD-MA to two recent datasets that measure transcriptome and chromatin accessibility in ~2000 single cells. To scale the runtime of MMD-MA to a more substantial number of cells, we extend the original implementation to run on GPUs. We also introduce a method to automatically select one of the user-defined parameters, thus reducing the hyperparameter search space. We demonstrate that the proposed extensions allow MMD-MA to accurately align state-of-the-art single-cell experiments.
|
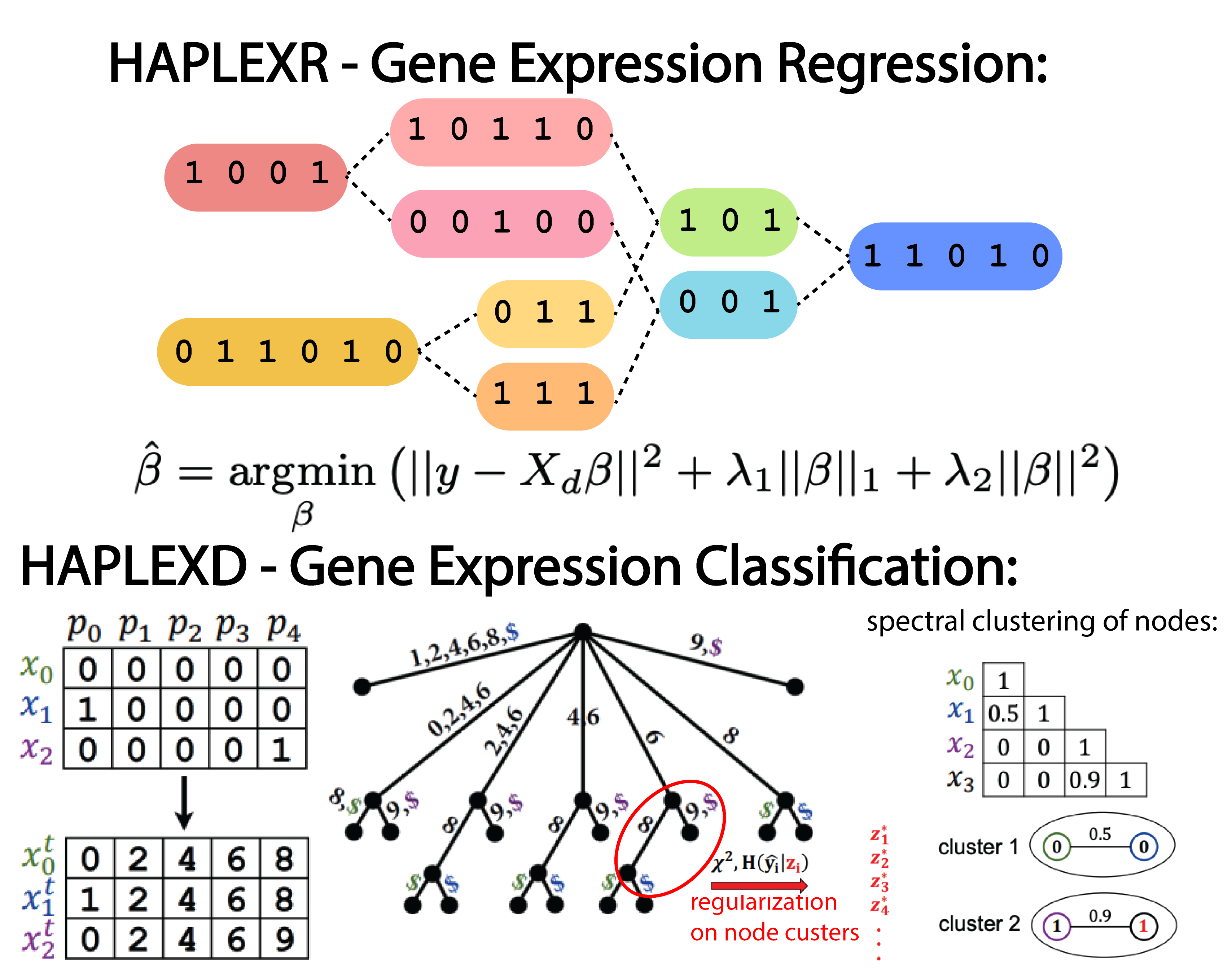 |
Combinatorial and statistical prediction of gene expression from haplotype sequence
B. Alpay*, P. Demetci* Sorin Istrail, and Derek Aguiar#
*Equal Contribution, #Corresponding Author
Bioinformatics (Oxford Press) vol.36, Supplement_1, p:i194-i202. 2020
Proceedings of the 27th International Conference on Intelligent Systems for Molecular Biology
(ISMB 2020)
[3]
[abstract] [paper] [code]
Genome-wide association studies (GWAS) have discovered thousands of significant genetic effects on disease phenotypes.By considering gene expression as the intermediary between genotype and disease phenotype, eQTL studies have interpreted many of these variants by their regulatory effects on gene expression. However, there remains a considerable gap between genotype-to-gene expression association and genotype-to-gene expression prediction. Accurate prediction of gene expression enables gene-based association studies to be performed post-hoc for existing GWAS, reduces multiple testing burden, and can prioritize genes for subsequent experimental investigation.In this work, we develop gene expression prediction methods that relax the independence and additivity assumptions between genetic markers. First, we consider gene expression prediction from a regression perspective and develop the HAPLEXR algorithm which combines haplotype clusterings with allelic dosages. Second, we introduce the new gene expression classification problem, which focuses on identifying expression groups rather than continuous measurements; we formalize the selection of an appropriate number of expression groups using the principle of maximum entropy. Third, we develop the HAPLEXD algorithm that models haplotype sharing with a modified suffix tree data structure and computes expression groups by spectral clustering. In both models, we penalize model complexity by prioritizing genetic clusters that indicate significant effects on expression. We compare HAPLEXR and HAPLEXD with three state-of-the-art expression prediction methods and two novel logistic regression approaches across five GTEx v8 tissues. HAPLEXD exhibits significantly higher classification accuracy overall; HAPLEXR shows higher prediction accuracy on approximately half of the genes tested and the largest number of best predicted genes ($r^2>0.1$) among all methods.
We show that variant and haplotype features selected by HAPLEXR are smaller in size than competing methods (and thus more interpretable) and are significantly enriched in functional annotations related to gene regulation. These results demonstrate the importance of explicitly modelling non-dosage dependent and intragenic epistatic effects when predicting expression.
|
2019
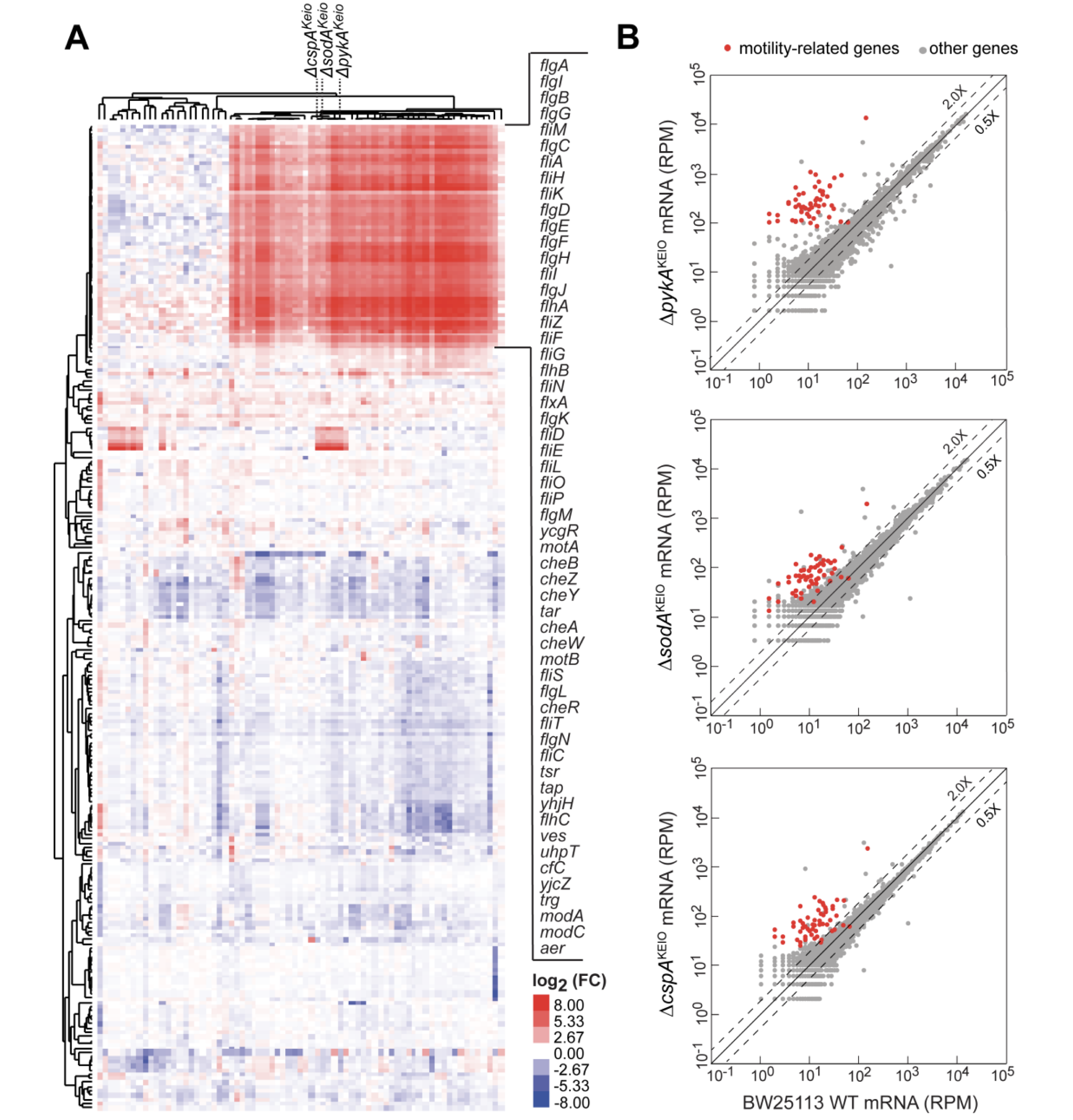 |
Rapid accumulation of motility-activating mutations in resting liquid culture of Escherichia coli
D. Parker*, P.Demetci*, and G.W. Li#
*Equal Contribution, #Corresponding Author
Journal of Bacteriology, 2019
[2]
[abstract] [paper]
Expression of motility genes is a potentially beneficial but costly process in bacteria. Interestingly, many isolate strains of Escherichia coli possess motility genes but have lost the ability to activate them in conditions in which motility is advantageous, raising the question of how they respond to these situations. Through transcriptome profiling of strains in the E. coli single-gene knockout Keio collection, we noticed drastic upregulation of motility genes in many of the deletion strains as compared to its weakly motile parent strain (BW25113). We show that this switch to a motile phenotype is not a direct consequence of the genes deletec, but is instead due to a variety of secondary mutations that increase the expression of the major motility regulator, FlhDC. Importantly, we find that this switch can be reproduced by growing poorly motile E. coli strains in non-shaking liquid medium overnight but not in shaking liquid medium. Individual isolates after the non-shaking overnight incubations acquired distinct mutations upstream of the flhDC operon, including different insertion sequence (IS) elements and, to a lesser extent, point mutations. The rapid sweep in the non-shaking population shows that poorly motile strains can quickly adapt to a motile lifestyle by genetic rewiring.
|
2016
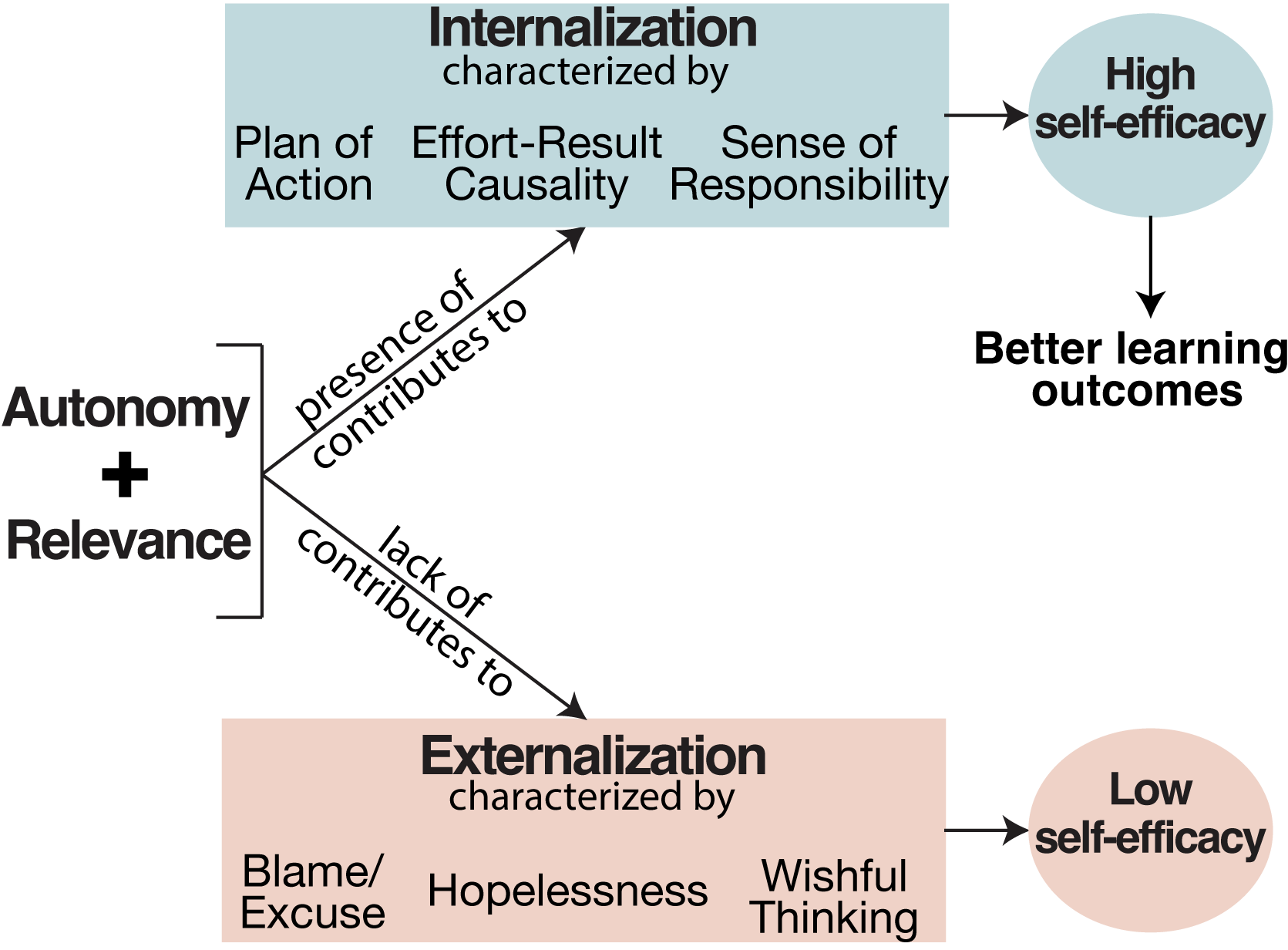 |
Internalization and externalization in the classroom: How do they emerge and why is it important?
P.Demetci, C. Nichols, Y. V. Zastavker, J. D. Stolk, A. Dillon, M. D. Gross.
IEEE Frontiers in Education
(FIE 2016)
[1]
[abstract] [paper]
“I felt so dumb, and it’s not fair that I cannot grasp this information to save my life, and other people can with no problem.” Why do some students feel empowered in the classroom, and feel they have control over their own learning, while others do not? Our qualitative investigation is a part of a larger mixed-methods study about students’ situational motivations in introductory STEM courses. We used grounded theory to analyze students’ responses to surveys about emotion, course relevance, and motivation. We investigated two emergent phenomena we called “internalization” and “externalization.” Our definitions of these are based on a student’s perception of who or what influences the outcomes of their activities: the students themselves, or external factors such as the instructor, peers, or the educational system as a whole. Our findings indicate that (1) internalization correlates with cognitive autonomy, students’ perception of course content having high personal relevance, and group projects in project-based learning environments; and (2) externalization correlates with lecture-based environments and students’ perceptions of lack of personal relevance in the course content. Our analyses suggest that project-based learning environments may serve to empower students, but only when course content is found to be relevant.
|
Awards and Honors
Last updated on 23 Jan 2024. Template based on github.com/bamos/bamos.github.io

 Pinar Demetci
Pinar Demetci